在复杂的有机分子(特别是天然产物萜烯)上选择性地进行C-H键转化是一种引入新官能团/偶联片段的有效方法。然而,因为烯丙基C-H键较弱,要想实现复杂底物的C-H键胺化反应,必须要控制位点选择性。此外,C=C 键的高反应活性也增加了该反应的难度(图1B)。目前,现有的方法都是基于氮宾或自由基的C-H键胺化反应,这会导致竞争性的叠氮化或烯烃加成反应,而不是烯丙基胺化反应。采用-烯丙基络合物可以避免叠氮化反应,但很容易发生烯烃双键的移位,因此难以控制反应的区域选择性,底物的适用范围较窄。此外,对这些反应体系进行优化也只能引入单一的氮取代基,或者需要特殊的氮源,因此存在一定的局限性。
1976年,Sharpless等人报道了化学计量硒试剂(由无水TsNClNa制备而成)参与的简单烯烃的烯丙基C-H键胺化反应( J. Am. Chem. Soc. , 1976, 98 , 269-271; Liebigs Ann. Chem ., 1975, 1725–1731; Angew. Chem. Int. Ed. Eng ., 1996, 35 , 454-456),但是反应的收率较低、氮源易爆炸且需要化学计量的硒试剂。于是,美国 华盛顿大学化学系的 Forrest E. Michael教授课题组设想能否使用催化量的硒试剂实现复杂底物萜类化合物的烯丙基C-H键胺化反应(图1A)?近日,他们在 J. Am. Chem. Soc. 上报道了一种 新型的无金属参与的烯烃烯丙基胺化反应,仅需催化量的硒试剂(www.58yuanyou.com硒化膦或硒脲),就可www.58yuanyou.com以在烯烃的烯丙基位引入各种含氮官能团。该反应不仅具有较高的收率和优异的区域选择性,还能用于萜类化合物的选择性胺化。
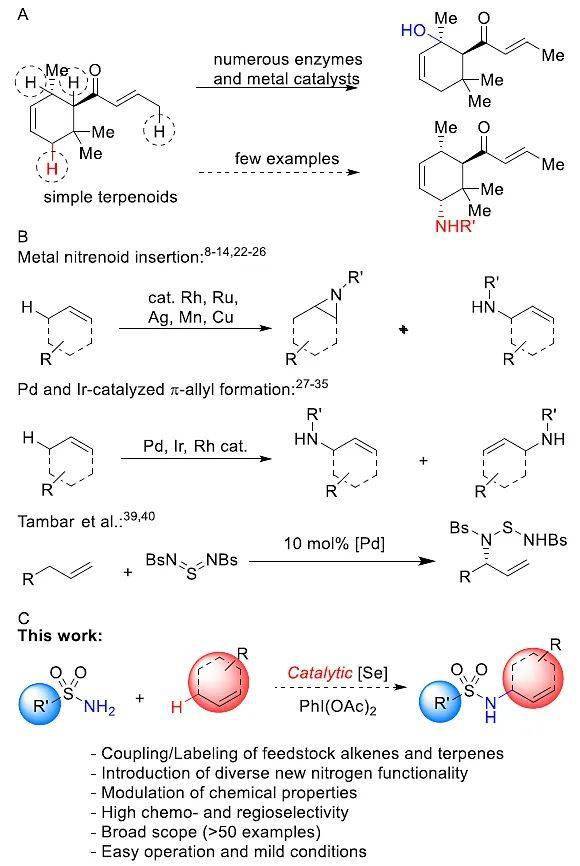
图1. 萜类化合物的C-H键胺化反应。图片来源: J. Am. Chem. Soc.
首先,作者在催化量硒粉和当量氧化剂PhI(OAc) 2 的存在下,使4-硝基苯磺酰胺(NsNH 2 )与烯烃(4-苯基-1-丁烯, 1)进行反应,结果并没有得到偶联产物。在过渡金属催化中,膦和 N -杂环卡宾(NHC)配体通常用于提高活性金属中心的溶解度和稳定性并控制其反应性。受此启发,作者认为使用带有合适配体的硒源(如硒化膦、硒脲)或许更有效。令人欣慰的是,硒化膦催化剂确实能够得到所需的烯丙基胺化产物,其中15 mol%Cy 3 PSe是最有效的催化剂。当催化剂的量降低至5 mol%时,收率仅稍有原由网下降。此外,在反应过程中没有检测到任何烯烃双键移位的产物,也没有观察到苄位的竞争性胺化产物。
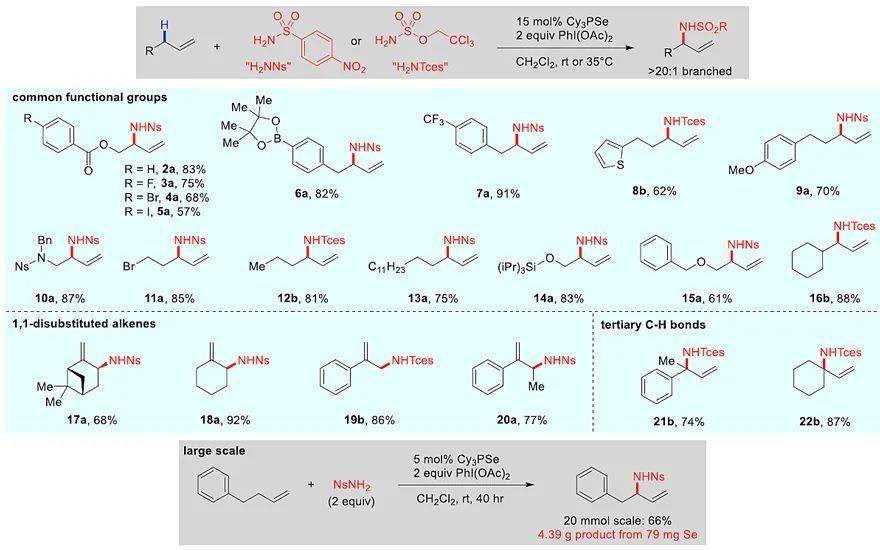
图2. 端烯底物范围。图片来源: J. Am. Chem. Soc.
在此基础上,作者考察了各种端烯和1,1-二取代烯烃(图2),结果显示该反应能够兼容各种官能团(如酯、保护的醇和胺、富电子芳族化合物、芳基硼酸酯以及烷基/芳基卤化物)。带有保护基的高烯丙基醇和高烯丙基胺也能实现这一转化,得到合成上十分有用的1,2-氨基醇和1,2-二胺。最重要的是,使用该方法可以有效地实现一级、二级和三级C-H键胺化反应。此外,该反应还能以克级规模进行,仅需5 mol%Cy 3 PSe(79 mg Se),就能生成4.39g产物 1a。值得注意的是,最容易脱除的两个胺保护基(4-硝基ySkewnG苯磺酰基(Ns)和2,2,2-三氯乙氧基磺酰基(Tces))均能以高收率得到目标产物。
随后,作者考察了磺酰胺和氨基磺酸酯的底物范围(图3)。几种合成上有用的胺保护基(Ns、Tces、Tfe、Nbos)都能以很高的收率(86-94%)实现胺化。此外,还可以引入后续进行偶联反应的基团,例如:交叉偶联反应的芳基卤化物/硼酸酯( 1e、 1f)、铜催化环加成反应的炔烃和叠氮化物( 1g、 1h)。值得一提的是,烯烃底物的极性/溶解度可通过胺化偶联体(如带有强酸性N-H键的TfNH( 1i)、长烷烃链( 1j)和全氟化碳链( 1k))进行修饰。此偶联步骤也可用于标记带有荧光基团的底物( 1l)。
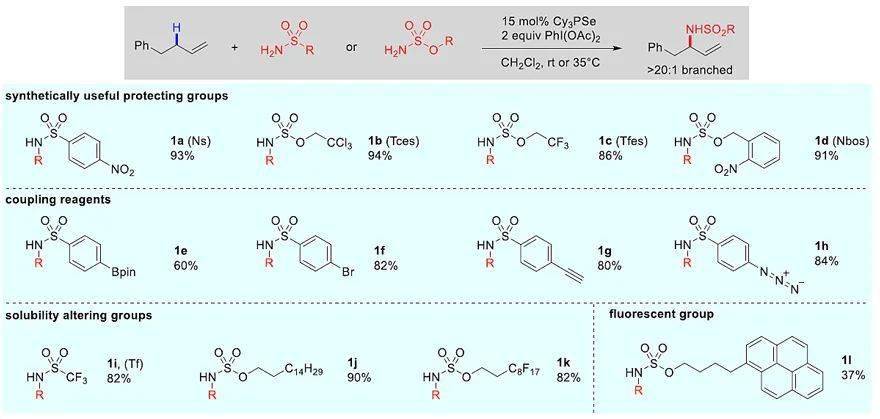
图3. 用功能性磺酰胺/氨基磺酸酯标记烯烃。图片来源: J. Am. Chem. Soc.
接下来,作者考察了多取代烯烃的底物范围,但结果不让人满意,反应的收率较低。于是,他们对配体进行筛选,发现只需将催化剂改为氮杂环卡宾衍生的硒脲IMeSe,就能使更富电子的烯烃获得高收率。在此基础上,作者考察了各种萜类化合物(图4),从单取代到四取代的每个烯烃都能以良好的产率获得烯丙基胺化产物,并且大多数情况下得到单一的区域异构体。对于链状三取代烯烃(如衍生自香茅醇和香叶醇的底物),胺化反应在烯基取代基较多的末端进行,以中等至较好的收率(60-91%)得到单一的烯基立体异构体( 23a-29a),且与第三个取代基呈反式。对于环状三取代烯烃底物而言,通常得到环内取代的胺化产物( 32a-34a),特别是在-紫罗兰酮( 33a)上高度拥挤的叔碳位进行胺化。如果环内位点被占据(如胆固醇、茶螺烷、神经醇和蒎烯),则会在环外进行胺化反应( 31a、35a-38a)。此外,最具挑战性的是带有环外异丙烯基的底物( 42b-45a),尽管反应的总收率较高,但是CH 3 和CH之间的选择性却较低。值得注意的是,该反应中观察到的区域选择性不同于在氮宾、自由基和-烯丙基胺化反应中所观察到的区域选择性,后者通常在烯烃取代基较少的末端进行胺化。
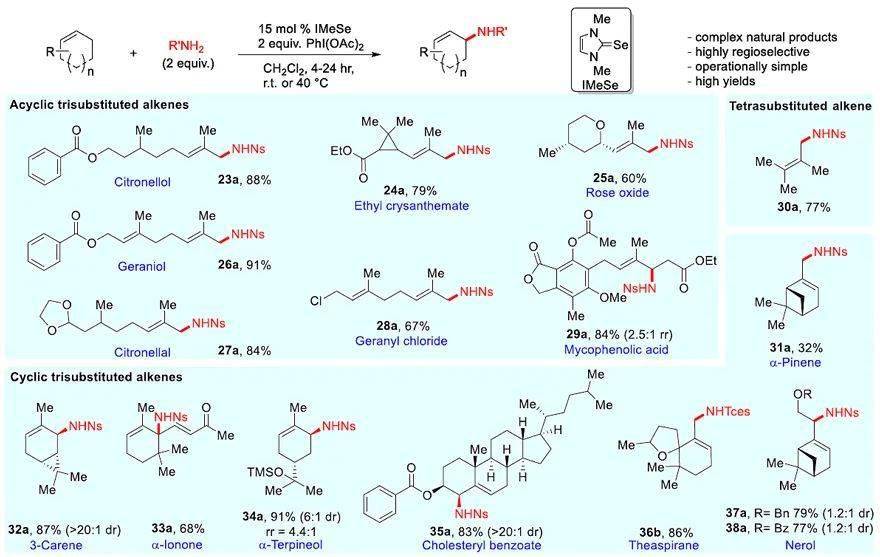
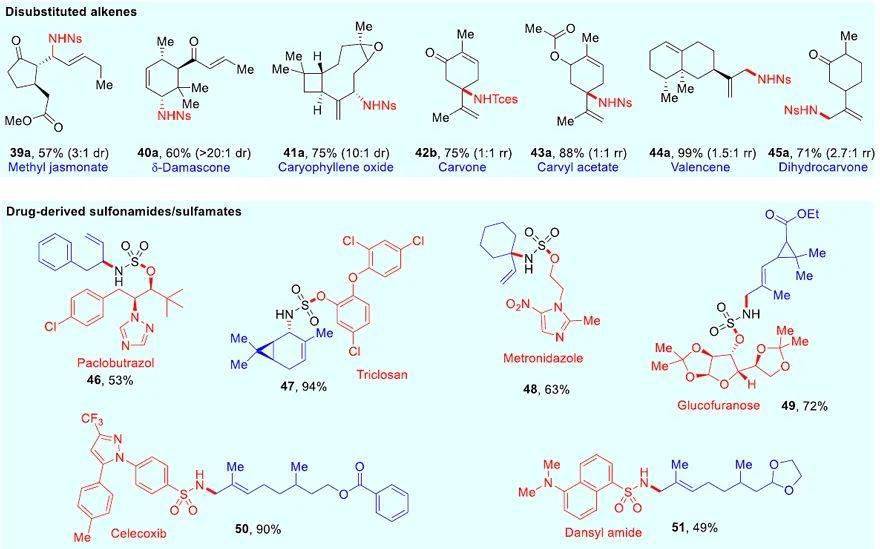
图4. 萜类化合物的胺化以及生物活性磺酰胺和氨基磺酸盐的标签。图片来源: J. Am. Chem. Soc.
为了进一步证明该反应的实用性,作者将其用于生物活性分子的修饰中ySkewnG(图4)。将衍生自生物活性分子的氨基磺酸酯和磺酰胺与不同的烯烃进行反应,以中等至较好的收率(53-94%)生成烯烃/药物共轭物(alkene/drug conjugates, 46-49)。此外,磺胺类止痛药塞来昔布( 50)和荧光基丹磺酰胺( 51)也能以较高的收率偶联到萜类化合物上。
如图5A所示,作者进行了一系列实验以验证配体在该反应中的作用,结果显示单独添加商业硒粉或Cy 3 P,反应无法进行;将Se和Cy 3 P分别添加到反应混合物中确实能够得到所需的产物,但同时也会生成Cy 3 PSe。事实上,在添加其他试剂前,将Se和Cy 3 P预混合仅5 min便可得到与预制Cy 3 PSe几乎相同的产率。此外,反应混合物的原位 31 P NMR谱显示Cy 3 PSe的信号立即消失,同时伴随着两个新物种的生成(图5B)。这两个新物种在反应过程中持续存在,且在有/无烯烃 1的情况下获得相同的NMR光谱。
鉴于目前尚未报道过双(酰亚胺)硒的分离或表征,因此作者将着重研究活性胺试剂(图5C),该试剂是由Sharpless在Et 3 N存在下通过SeCl 4 和TsNH 2 制备而成。将该混合物分别用当量的Cy 3 P、Cy 3 PSe或Cy 3 PO处理,得到相应的 31 P NMR谱。然后,将烯烃 1(1equiv)分别加入到这些混合物中反应24 h,结果显示添加Cy 3 P的 31 P NMR谱会出现三个新的峰,且都不对应于游离Cy 3 P。作者将它们分别指认为Cy 3 PNTs(38 ppm)、Cy 3 PO(49 ppm)和Cy 3 PSe(58 ppm),这表明游离的膦被假定的硒双(酰亚胺)快速氧化,并且在反应过程中不可能持续存在。将Cy 3 PSe添加到硒双(酰亚胺)中,Cy 3 PSe的 31 P NMR不变。但是,向此混合物中添加Cy 3 PO会导致甲苯磺酰基的 1 H NMR谱发生显著的变化,同时Cy 3 PO 的 31 P NMR谱移至52 ppm。值得注意的是,向假定的游离硒双(酰亚胺)中添加Cy 3 P和Cy 3 PSe,烯烃 1生成胺化产物 1m的收率较低。但是,添加Cy 3 PO可使转化率显著提高(51%),且与标准条件下 1m的收率相同(51%)。
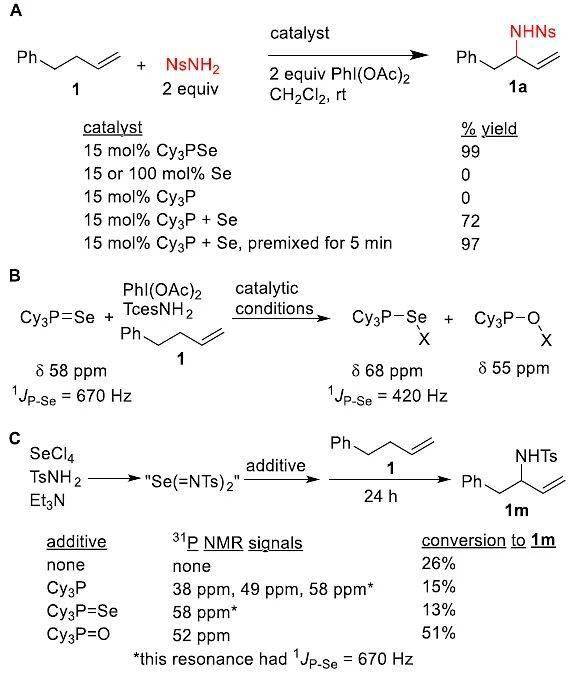
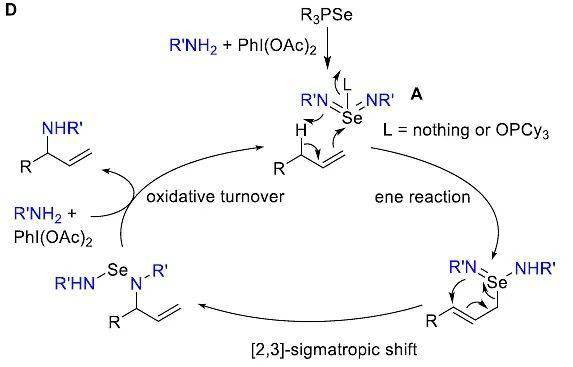
图5. 机理研究及可能的机理。图片来源: J. Am. Chem. Soc.
如图5D所示,作者提出了可能的反应机理。首先,Cy 3 PSe被PhI(OAc) 2 /RSO 2 NH 2 氧化为硒双(酰亚胺) A。其次, A与底物 1发生ene反应、[2,3]-sigmatropic迁移得到胺化中间体。最后,胺化中间体在PhI(OAc) 2 /RSO 2 NH 2 的氧化下得到产物,并再生硒双(酰亚胺) A。
总结
Forrest E. Michael教授课题组开发了一种新型的硒参与的烯烃烯丙基C-H键胺化反应。该反应条件温和、无需金属催化、反应收率好、区域选择性高、底物适用性广(从单取代到四取代的烯烃都能兼容),并可用于各种萜类天然产物的选择性胺化反应,为制备生物活性分子提供了一条简单高效的策略。
Catalytic Metal-free Allylic C-H Amination of Terpenoids
Wei Pin Teh, Derek C. Obenschain, Blaise M. Black, Forrest E. Michael
J. Am. Chem. Soc., 2020, DOI: 10.1021/jacs.0c06997
导师介绍
Forrest E. Michael
https://www.x-mol.com/university/faculty/1599
本文版权属于 X-MOL(x-mol.com),未经许可谢绝转载!欢迎读者朋友们分享到朋友圈or微博!







